National Seed Health System
ARCHIVED 08.03.2017 Cb 1.3 – Acidovorax avenae ssp citrulli – Syngenta SYBR PCr
| VERSION: 1.0 |
| DATE: [ARCHIVED 08.03.2017] |
| PATHOGEN: Acidovorax avenae ssp. citrulli (syn: A. citrulli) |
| HOST: Watermelon (Citrullus lanatus var. lanatus); Cantaloupe (Cucumis melo var. cantalupensis); Melon (Cucumis melo) |
| COMMON NAME: bacterial fruit blotch (BFB) |
| METHOD: Cb 1.3 Syngenta SYBR Green PCR Method |
| METHOD CLASS: Temporary Standard (B) |
| SAMPLE: 10,000 to 30,000 seeds |
PROCEDURE:
Sample Preparation
1. As a general lab cleanliness practice, ensure all pertinent laboratory surfaces have been properly cleaned to prevent contamination.
2. Weigh out appropriate number of subsamples of 5,000 seeds (2500 seeds for rootstocks) and place seeds in sample buckets. It is recommended that a 4 L bucket be used for larger seed such as squash and a 2.5 L bucket used for smaller seed such as watermelon. Record the 5,000 seed weight for further use.
3. Calculate and add the necessary amount of seed wash buffer (SWB).
a. For melons and watermelons: 2.0 ml x 5,000 seed weight (i.e., 400 ml for 200 g)
b. For rootstocks: 2.5 ml x 2,500 seed weight (500 mL for 200 g)
4. Spike with suspension of A. avenae subspecies cattleyae adjusted to 0.1 absorbance OD600 for final concentration of approximately 104 colony forming units/ml of buffer or adding a sufficient volume of a frozen stock in 10-15% glycerol suspension to achieve 104 colony forming units/ml of buffer.
5. Submerge seeds and rotationally shake for 16-24 hours at 100 RPM at room temperature.
Bacterial Extraction
6. For each subsample, transfer 10.0 ml of seed wash solution to a 15 ml centrifuge tube (VWR #21008-212 or similar product). Seed wash solution can be stored at -20°C after step 6 until further processing.
7. Centrifuge at 1200 RCF for 5 min.
8. Transfer the supernatant to a new 15 ml centrifuge tube and centrifuge at 3220 RCF for 15 min.
9. Taking care not to disturb the pellet, decant the supernatant.
10. Resuspend the pellet in 1.0 mL of sorbitol solution and incubate in a fume hood at room temperature for 20 min (maximum 1 hr).
11. Centrifuge for 10 min at 1800 RCF, decant the supernatant, and proceed to step 12 if extracting
18 or fewer subsamples or to step 38 if using the 96-well extraction equipment.
DNA Extraction for low output extractions
12. Resuspend the pellet in 750 µL PBS and transfer to a sterile microcentrifuge tube.
13. Centrifuge for 3 min at maximum RCF in the microcentrifuge.
14. Turn on water bath and set for 65°C.
15. Use the Machery-Nagel (MN) NucleoSpin Plant II (Art-Nr: 740770.250) for low output extractions.
16. Make a Lysis Buffer stock of 480 µL buffer MN PL1 plus 20 µL Proteinase K (20 mg/mL) per extraction.
17. Pour off supernatant and resuspend in 450 µL of Lysis Buffer.
18. Mix thoroughly with pipette and vortex.
19. Vortexing the samples every 15 min, incubate for 1 h at 65°C.
20. Label clean microcentrifuge tubes and add 450 µL of buffer PC to them.
21. Upon removal, change temperature of water bath to 70°C.
22. In microcentrifuge, centrifuge samples for 5 min (maximum RCF) to clear the lysate.
23. Do not disturb pellet. Add 400 µL of the cleared lysate to the PC buffer; vortex thoroughly.
24. Load 700 µL of the sample into a NucleoSpin Plant II Column (green ring).
25. In microcentrifuge, centrifuge for 1 min (maximum RCF) and discard flow-through.
26. Add the remainder and spin.
27. Discard flow-through and add 400 µL PW1 to the column and centrifuge for 1 min (maximum RCF); discard flow-through.
28. Repeat step 26 and 27 once.
29. Place PE buffer into water bath.
30. Discard flow-through and add 700 µL PW2 to the column.
31. In microcentrifuge, centrifuge for 1 min (maximum RCF).
32. Discard flow-through and add 200 µL PW2 to the column.
33. Centrifuge for 2 min (maximum RCF) to dry the membrane.
34. Place column into new labeled microcentrifuge tube.
35. Add 100 µL of pre-heated PE buffer directly to column membrane; incubate for 5 min at 70°C.
36. Centrifuge for 1 min at maximum RCF.
37. Proceed to PCR amplification at step 73.
DNA Extraction for high output extractions
38. Resuspend the pellet in 500 µL PBS.
39. Transfer suspensions to a 0.8 mL deep well plate with V-bottom (Thermo Scientific AB 0859) for further work in 96-well format.
40. Cover the 96-well plate with Scapa tape or similar used for ELISA.
41. Centrifuge the suspensions in the deep well plate at 3220 RCF for 15 minutes.
42. Decant the supernatant and dab the deep well plate on a tissue after so no contamination can occur.
43. With forceps or dispenser, add three 3 mm diameter glass beads (Walter Stern 100C, Port Washington, NY or similar product) to each well.
44. Turn on water bath and set for 65°C.
45. Use the MN NucleoSpin 96 Plant II (Art-Nr: 740663.24) for high throughput, 96-well format extractions.
46. As a general lab cleanliness practice, ensure all pertinent laboratory surfaces have been properly cleaned with DNase I (Thermo Scientific) or other product(s) with similar nucleic acid activity to prevent contamination.
47. Add 450 µl lysis buffer [per 480 µl PL1 buffer + 20 µl Proteinase K (20 mg/ml)] to each sample well. Prepare at least one negative control during DNA isolation using only the buffers provided in the kit.
48. Seal the plate with aluminum seal (Thermo Scientific Easy Peel cat. # AB-0745) using 4titude model S120499-1-A004 or equivalent. Ensure the top of the wells is dry to facilitate sealing. (Note: this is important as covering with plastic performs poorly during the 65°C incubation).
49. Resuspend the pellets by inverting the plate several times. If not resuspended well, place the plate in the Qiagen TissueLyser and shake for 10 seconds (0.10 min on the device) at 20.0 Hz (20.0 1/s on the device).
50. Incubate the suspension for 1 hour at 65°C in the shaking incubator.
51. Centrifuge at up to 5,000 RCF for 20 minutes to clear the lysate.
52. In the meantime, add 450 µl PC buffer to the needed wells in a 2.2 mL deep well plate (from the Macherey Nagel kit with square wells) or microcentrifuge tubes.
53. Place the spacers ‘MTP/Multi-96 plate’ and the receptacle into the vacuum manifold.
54. Put an MN Wash Plate on the spacer. Failure to place on spacer can result in cross contamination.
55. Close the vacuum manifold and place a NucleoSpin Plant II binding plate on the manifold.
56. Cover the unused wells with laboratory tape.
57. Pipette 400 µl of cleared lysate to the PC buffer. Note: Do not disturb the pellet during pipetting.
58. Mix the solution well a few times by pipetting. Note: Pipette slowly to prevent foaming and cross contamination.
59. Load the sample on the NucleoSpin Plant II binding plate.
60. Turn on vacuum (-200 mbar) and make sure the flow-through rate is not faster than 1-2 drops per second.
61. Pipette 400 µl of Buffer PW1 into each well and put in vacuum (-400 mbar).
62. Turn off the vacuum once all the buffer is pulled through the column and air vent.
63. Repeat above two steps once.
64. Pipette 700 µl of Buffer PW2 into each well and turn on vacuum (- 400 mbar).
65. Turn off the vacuum once all the buffer is pulled through the column and the air vent.
66. Remove the Wash MN Plate and receptacle from the vacuum manifold.
67. Dry the membrane in the wells by vacuum at full strength for 15 minutes.
68. Turn off the vacuum.
69. Add 100 µl of warmed (70°C) PE buffer in the wells or pillars.
70. Incubate for 2 minutes at room temperature.
71. Turn on vacuum (full vacuum, ~ 600 mbar) until samples have gone through the columns.
72. Proceed to PCR amplification at step 73.
PCR Amplification
73. Prepare the PCR mix for real-time PCR thermocycler in an ethanol cleaned and DNase treated laminar flow or PCR workstation.
74. A master mix for all reactions should be made to reduce the effect of pipetting errors of small quantities.
75. Template from each of the subsample extractions should be added to a reaction tube.
76. For each subsample, two reactions at each template concentration should be conducted.
77. Use 10.0 µL of the extracted DNA as the PCR template so the total reaction volume is 25.0 µL.
78. One reaction per PCR run should be prepared and spiked with extracted Aacat DNA to serve as a control for the PCR run and to determine the melt peak for the Aacat PCR product.
79. One reaction per PCR run should be prepared and spiked with extracted Aac DNA as the template to serve as a positive control for the PCR run. To prevent the chance of contamination, this reaction should be the last template to be thawed and added.
80. Proceed with the PCR using the thermocycler conditions listed below. Record the location of the samples in the thermocycler cells.
81. After the PCR run, a dissociation curve is conducted after the completion of PCR to confirm the product. Begin at 65°C and proceed at 0.5°C per second up to 95°C.
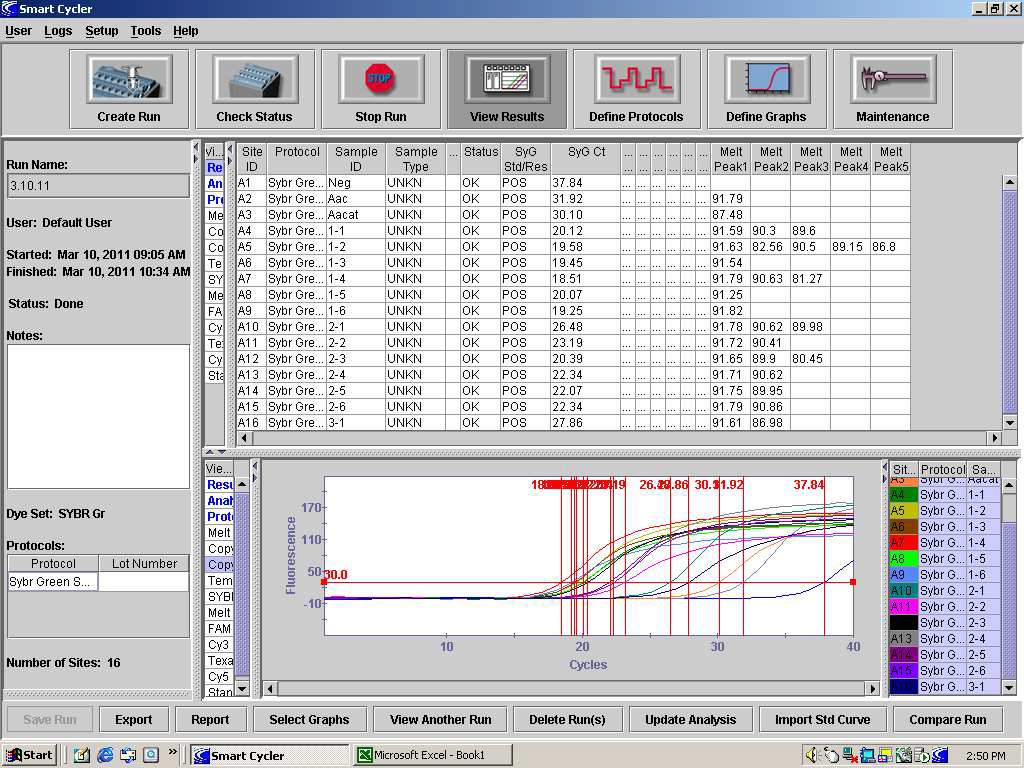
82. After completion of dissociation, detection of the internal control should occur at approximately 88°C and detection of Aac should occur, if present, at approximately 91°C. When analyzing the results of the PCR, the temperature of the Aacat melt peak in the control reaction should be consulted to determine if the extraction and PCR are valid. See Figure 1. Product dissociation temperature can be different among thermocyclers so use the PCR controls to provide temperature data for such comparisons.
83. Undiluted samples with CT values over 31 32 or with a failure to detect the internal control should be subjected to PCR again. In this case, the DNA template should be diluted 10-1 and 10-2 to adequately reduce the DNA quantity for PCR to be performed.
84. Reactions with DNA products having a melt peak at approximately 91°C should be suspect of having active and virulent Aac and should be considered for either disposal or tested using a bioassay.
PCR reaction mixture per reaction
SYBR Green (BioRad) 12.5 µL
ZUP 2436 (20 µM) 0.5 µL
ZUP 2437 (20 µM) 0.5 µL
ZUP 2424 (20 µM) 0.5 µL
ZUP 2425 (20 µM) 0.5 µL
sterile PCR grade water 0.5 µL
DNA template 10.0 µL
Primer specifications:
| primer | sequence | length | target | product size (bp) |
|---|---|---|---|---|
| ZUP2424 ZUP2425 ZUP2436 ZUP2437 |
5’- CACGCCTCTAGCCAACAAC -3’ 5’- GATGCTCACAATCTCGGACC-3’ 5’- AAGGCGTTTTTGTAGCCAGA-3’ 5’- GAACTGCTGCACATCGACAC-3’ |
19 20 20 20 |
Aacat: control Aacat: control Aac: routine Aac: routine |
183 183 281 281 |
Thermocycler conditions
1. 180 seconds at 95°C
2. 30 seconds at 95°C
3. 30 seconds at 61°C
4. 45 seconds at 72°C
40 cycles of step 2 to step 4
RECIPES:
5x Phosphate Buffer Solution (PBS):
NaCl 40.0 g
KH2PO4 1.0 g
KCl 1.0 g
Na2HPO4*12H2O 14.5 g
deionized water 1000 mL
Adjust pH to 7.4 prior to autoclaving
Phosphate Buffer Solution + Tween 20 (PBST):
PBS 5x 200 mL
Tween 20 (Sigma-Aldrich, P1379) 0.5 mL
deionized water 800 mL
Seed Wash Buffer (SWB):
PBS 5x 200 mL
Tween 20 (Sigma-Aldrich, P1379) 0.5 mL
Nystatin (Sigma-Aldrich, N6261) 20 mg
deionized water 800 mL
Sorbitol Solution:
D-sorbitol (Sigma-Aldrich) 0.81 g
1M Tris-HCl pH 8.0 (Invitrogen) 1.30 ml
0.5M EDTA pH 8.0 (Invitrogen) 130 µL
β-mercaptoethanol 98% (Sigma-Aldrich M7154) 26 µL
sterile deionized water 11.7 mL
The above recipe is for a 13 mL final volume.
FIGURE 1: The dissociation analysis of the PCR products from a Syngenta SYBR Green PCR run of
two positive seed lots